Dr. Grattan's research interest involves the synthesis and development of improved enzyme inhibitors for cancer research.
SC-INBRE Research
Design of Novel Inhibitors of Human Sphingosine Kinases 1 and 2
Sphingolipids are a family of compounds
that, in addition to being structural constituents of cell membranes, play key
roles as signaling molecules. In
particular two of these sphingolipid metabolites, ceramide and sphingosine
1-phosphate (S1P), have recently received considerable attention as integral
mediators of cell death and survival. The regulator of the ceramide/S1P equilibrium is sphingosine kinase-1
which phosphorylates sphingosine to form S1P. Sphingosine kinase-1 has been identified
as an oncogene and is, therefore, of considerable interest in the treatment of
cancer. To this end, a number of
novel inhibitors of sphingosine kinase-1 have recently been identified and
evaluated by Smith et al. These inhibitors
show promising chemotherapeutic results in vitro, but are simply a starting
point in the eventual optimization of in vivo activity. Work has recently begun, in
collaboration with Smith's lab, on developing a synthetic route to produce one
of these inhibitor compounds as a template molecule. The design and ultimate completion of
this synthetic scheme will allow for numerous derivatives to be synthesized
quickly and concisely in effort to evaluate and increase the therapeutic effect
of sphingosine kinase-1 inhibition.
Considered the central molecule in
sphingolipid metabolism, ceramide controls the programmed cell death response
to a wide array of anticancer treatments through de-novo synthesis and/or the
hydrolysis of sphingomyelin.
1 Typical treatments, such as chemotherapy and radiation, elicit an
increase in the intracellular ceramide level occurring before the first
biochemical signs of apoptosis.
1a The addition of extracellular
short-chain ceramides to cell culture results in apoptosis for a number of
cancer cell lines.
1a In
contrast to ceramide, S1P promotes cell survival in response to the apoptotic
stresses that typically induce ceramide generation
in vitro,
ex vivo, and
in vivo.
2 The opposing directions of
ceramide-mediated and S1P-mediated signaling led to the concept of a
ceramide/S1P biostat, and the assumption that the ratio between these two
lipids ultimately determines the fate of the cell.
2a Both of these metabolites, ceramide and
sphingosine, have been associated with apoptosis and growth arrest in response
to multiple stress signals, while simultaneously increasing sphingosine kinase
activity as a prosurvival response. This increase in S1P levels may also be regulated through enhanced S1P phosphatase and S1P lyase activities as shown in Figure 1.
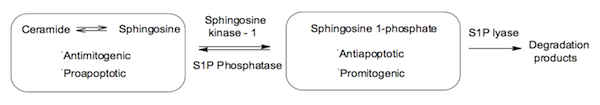
Figure 1. The sphingolipid biostat
Since the discovery that S1P regulates cell growth
3
and suppresses apoptosis
4, there have been numerous important
physiological and pathophysiological processes reported to be managed by S1P in
higher organisms. To further
highlight its importance as a signaling molecule, S1P has also been shown to
regulate biological responses in lower organisms. The activity of sphingosine kinase,
which exclusively catalyzes the ATP-dependent phosphorylation of sphingosine,
is stimulated by many pathways. Sphingosine kinases, SphK1 and SphK2, have been recently found to be
expressed in humans, mice, yeast and plants with homologues in worms and flies
and each with five conserved domains (Figure 2). The distinctive catalytic domain
contained with C1-C3, and the ATP binding site being identified within C2. These two isoforms do exhibit differences
in terms of the presence of transmembrane (TM) regions, SphK1 has none while SphK2
has four TM regions. The sequence
differences between these two proteins have led researchers to conclude that
they are the result of separate gene-duplication events. SphK1 and SphK2 have been cloned and
characterized in mammals.
5 Diverse external stimuli, particularly growth and survival factors,
stimulate SphK1, generating S1P that has been implicated in their mitogenic and
anti-apoptotic effects.
6,7 In contrast to SphK1, rather than promoting growth and survival,
overexpression of SphK2 suppressed growth and enhanced apoptosis,
8,9
implying that they have distinct physiological functions, likely due to their
different subcellular localizations. Northern blot analysis has shown that SphKs have different tissue
distributions
10:
SphK1 expression is highest in lung and
spleen, while SphK2 is more abundant in liver and heart. SphK1 has also been identified as the
key enzyme in modulating ceramide and sphingosine 1-phosphate levels, as shown
in Figure 1, and is therefore the focal point of our project.
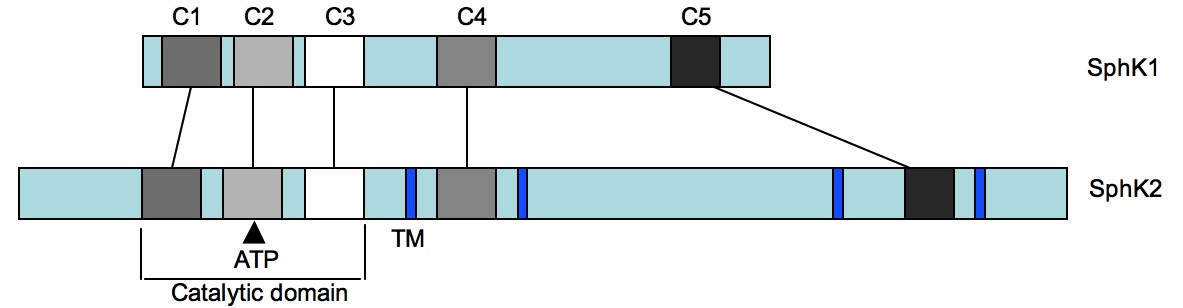
Figure 2. Isoform comparison of sphingosine kinase
1 and sphingosine kinase 2
During a recent screening of a synthetic compounds
library using an assay designed for recombinant human sphingosine kinase
activity testing, Smith
et al. identified
a new panel of SphK inhibitors.
11a These compounds, I-IV, as well as one
synthetic derivative Compound V, (Figure 3) have been found to possess
selectivity towards SphK in comparison with other lipid and protein kinases and
are not competitive inhibitors of the ATP-binding site of SphK. These nonlipid-based compounds
demonstrated activity at sub-micromolar concentrations, making them more potent
than any previously reported SphK inhibitor. The inhibitors are also
antiproliferative toward a panel of human tumor cell lines with simultaneous
induction of apoptosis. The
compounds inhibit S1P formation in intact cells and maintain activity toward
cells that express the drug transport proteins P-glycoprotein (Pgp) or
MRP1. Overall, a series of potent,
structurally novel lead inhibitors of SphK were identified and due to the
antiproliferative potential as drugs a synthetic scheme to design and
subsequently evaluate these derivatives was required.
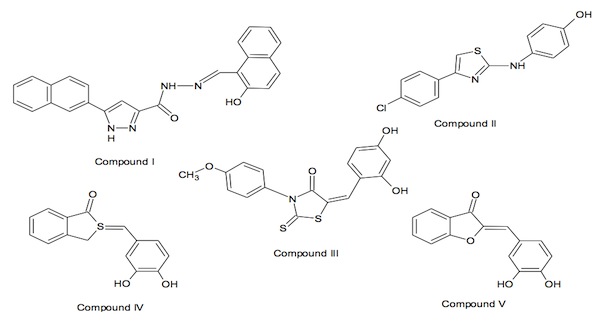
Figure 3. Novel non-lipid inhibitors identified by
Smith et al.
Compounds I, II, III, and IV (at 5
µg/ml) inhibited SphK activity by 99,
85, 99, and 89%, respectively, and serve as template structures for nonlipid
inhibitors.
11a Upon
evaluation of these structures, the chemotype of compound IV was the most
easily synthesized of the four classes of compounds to quickly provide a
suitable test derivative. This
bioisosteric replacement was expected to produce a compound with comparable
activity and resulted in compound V.
11a
A common problem with known kinase inhibitors is their
tendency toward nonselectivity because the majority of these inhibitors
interact with the highly conserved nucleotide binding site. Therefore, we performed competition
assays in which sphingosine and SK concentrations were held constant, whereas
ATP concentrations were varied. For
each inhibitor, the K
M for ATP and the
Vmax for
S1P formation was determined. Compounds that are competitive inhibitors for the ATP-binding site would
be expected to increase the K
M for ATP without affecting the
Vmax
of the reaction. The data for
compounds I–IV are summarized in Table 1.
11a The
Vmaxs show
significant decreases with all of the test compounds
versus vehicle
alone. In contrast, K
Ms were not increased such that ATP
concentrations up to at least 10 times the K
M were unable to
overcome inhibition by the compounds. Therefore, these compounds are not
competitive inhibitors at the ATP-binding site of SphK.
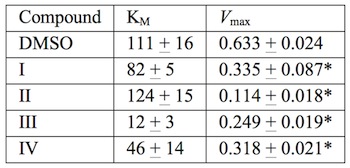
Table 1 Effects of SphK inhibitors on
Michaelis-Menten parameters for ATP
The effects of compounds
I–V were determined at multiple concentrations, and IC
50s for
each compound were calculated for human isoforms of ERK2, PI3k, and PKC-
α. GST-hSK inhibition data is
provided for comparison. The compounds demonstrated IC
50s
in the sub- to low micromolar range, making them more potent inhibitors of SphK
than any previously reported compound.
11a Biological evaluations demonstrated that
the IC
50s for inhibition of SphK and tumor cell proliferation by the
newly synthesized compound V, ~2
µM,
indicated that it is somewhat less potent than compound IV, ~0.6
µM.
In continuing studies of these SphK inhibitors as
cancer therapeutic agents, Smith reported additional in vitro and in vivo
properties of three SphK inhibitors (Compounds I, II and V).
11b Their findings show that the antitumor
activities mostly correlated well with their concentrations in blood and
tumors. When inhibitor
concentrations were normalized to equal S1P formation inhibition, the rank
order of modulation for both pathways was V > II >> I. The reason for differences in pathway
modulation is unknown. From our
previous kinase selectivity assays, it was observed that V was the most
promiscuous, potently inhibiting PI3k.
11a Therefore, compound V may potently
inhibit SphK but most likely inhibits other kinases as well. One of the
inhibitors, II, showed promising oral bioavailability and antitumor activity
while compounds I and V showed extremely poor oral bioavailability.
11b These findings provide additional
validation for sphingosine kinase-1 as a cancer therapeutic target, as well as
confirming that further evaluation of these small molecule inhibitors is
required.
There are four separate areas of
consideration in the currently accepted evaluation of whether a compound with
certain pharmacological or biological activity has the properties to become a
suitable drug candidate. The rule
describes molecular properties important for a drug's pharmacokinetics in the
human body, including their absorption, distribution, metabolism, and excretion
("ADME"). The rule is
integral for drug development where a pharmacologically active lead structure
is optimized step-wise for increased activity and selectivity, as well as
drug-like properties as described by Lipinski's rule.
12 The
modification of the molecular structure often leads to drugs with higher
molecular weight, more rings, more rotatable bonds, and a higher lipophilicity. However, the rule does not predict if a
compound is pharmacologically active.
To better understand the pharmacophoric
nature of Compound 1, we will design and synthesize various derivatives in an
attempt to improve upon the oral bioavailability of the inhibitors while
maintaining the overall activity.This will allow for more of an in-depth examination of this region's
role in the binding of these inhibitors to the target enzyme and allow for the
development of interesting organic methodology by the undergraduate student.
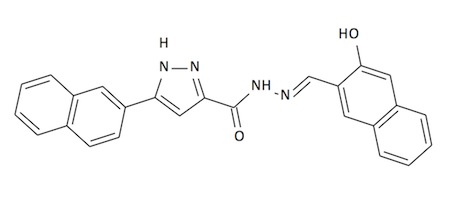
Compound 1
References
1. (a) Ogretman, B.; Hannun, Y. A.
"Biologically active sphingolipids in cancer pathogenesis and treatment,"
Nat. Rev. Cancer 2004,
4, 604. (b)
Senechenkov, A.; Litvak, D. A.; Cabot, M. C. "Targeting ceramide metabolism: a
strategy for overcoming drug resistance,"
J.
Natl. Cancer Inst. 2001,
93, 347.
2. (a) Kohama, T.; Olivera,
A.; Edsall, L. C.; Nagiec, M. M.; Dickson, R.; Spiegel, S. "Molecular cloning
and functional characterization of murine sphingosine kinase,"
J. Biol. Chem. 1998,
273, 23722. (b)
Liu, H.; Suguira, M.; Nava, V. E.; Edsall, L. C.; Kono, K.; Poulton, S.
et al. "Molecular cloning and functional
characterization of a novel mammalian sphingosine kinase type 2 isoform,"
J. Biol. Chem. 2000,
275, 19513. (c)
Johnson, K. R.; Becker, K. P.; Facchinetti, M. M.; Hannun, Y. A.; Obeid, L. M.
"PKC-dependent activation of sphingosine kinase 1 and translocation to the
plasma membrane. Extracellular release of sphingosine-1-phosphate induced by
phrobol 12-myristate 13-acetate (PMA),"
J.
Biol. Chem. 2002,
277, 35257.
3. (a) Zhang, H.
et al. "Sphingosine 1-phosphate, a novel
lipid, involved in cellular proliferation,"
J.
Cell Biol. 1991,
114, 155. (b) Olivera, A.; Spiegel, S.
"Sphingosine 1-phosphate as a second messenger in cell proliferation induced by
PDGF and FCS mitogens,"
Nature,
1993,
365, 557.
4. Cuvillier, O.
et al. "Suppression of ceramide-mediated
programmed cell death by sphingosine 1-phosphate,"
Nature,
1996,
381, 800.
5. Spiegel, S., and Milstien, S. "
Sphingosine-1-phosphate:
an enigmatic signalling lipid," Nat. Rev. Mol. Cell Biol., 2003,
4, 397–407.
6. Taha, T. A., Hannun, Y. A., and Obeid, L. M.
"Sphingosine kinase: biochemical and cellular regulation and role in
disease," J. Biochem. Mol. Biol., 2006,
39, 113–131.
7. Milstien, S., and Spiegel, S. "
Targeting
sphingosine-1-phosphate: A novel avenue for cancer therapeutics," Cancer Cell, 2006,
9, 148–150.
8. Okada, T., Ding, G., Sonoda, H., Kajimoto, T.,
Haga, Y., Khosrowbeygi, A., Gao, S., Miwa, N., Jahangeer, S., and Nakamura, S.
"Involvement of N-terminal-extended Form of Sphingosine Kinase 2 in
Serum-dependent Regulation of Cell Proliferation and Apoptosis
,"
J. Biol. Chem., 2005, 280,
36318–36325.
9. Maceyka, M., Sankala, H., Hait, N. C., Le Stunff,
H., Liu, H., Toman, R., Collier, C., Zhang, M., Satin, L., Merrill, A. H.,
Jr., Milstien, S., and Spiegel, S. "
SphK1 and
SphK2, Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid
Metabolism," J. Biol. Chem., 2005, 280, 37118–37129.
10. Liu, H., Chakravarty, D.,
Maceyka, M., Milstein, S., Spiegel, S. "Sphingosine kinases: a novel family of
lipid kinases,"
Prog. Nucleic Acid Res.
Mol. Biol. 2002,
71, 493-511.
11. (a) French, K. J.;
Schrecengost, R. S; Lee, B. D.; Zhuang, Y.; Smith, S. N.; Eberly, J. L.; Yun,
J. K.; Smith, C. D. "Discovery and evaluation of inhibitors of human
sphingosine kinase,"
Cancer Res. 2003,
63, 5962. (b) French,
K. J.; Upson, J. J.; Keller, S. N.; Zhuang, Y.; Yun, J. K.; Smith, C. D.
"Antitumor activity of sphingosine kinase inhibitors,"
J. Pharmacol. Exptl. Ther. 2006,
318, 596.
12. Lipinski, C. A., Lombardo, F., Dominy, B. W., Feeney, P. J. "
Experimental and computational approaches to
estimate solubility and permeability in drug discovery and development settings,"
Adv. Drug Del. Rev.,
2001,
46,
3-26.
Current Students
- Kevin Mays
- Amber Wallace
- Jaclyn Cika
Former Students
- Matt Wilson
- Erin White Wilson
- Demetrius Miles
- Madalyn McCaulley
- Nicole Quigley
- Valencia Fleming
- Ray Olang